
The unprecedented global healthcare crises caused by the outbreak of the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has driven extensive research focused on understanding the virus’ infection mechanism, which will help develop effective therapeutics.
Although previous studies have reported protein-protein interaction linkages during the viral infection life cycle, a more comprehensive understanding of the full interactome containing human micro ribonucleic acids (miRNAs), protein-coding genes, and co-infecting microbes is crucial.
 Study: Constructing a multiple-layer interactome for SARS-CoV-2 in the context of lung disease: Linking the virus with human genes and co-infecting microbes. Image Credit: Connect world / Shutterstock.com
Study: Constructing a multiple-layer interactome for SARS-CoV-2 in the context of lung disease: Linking the virus with human genes and co-infecting microbes. Image Credit: Connect world / Shutterstock.com
About the study
In a recent study published on the preprint server bioRxiv*, a team of researchers recently developed a statistical modeling method known as multiple-layer crosstalk (MLCrosstalk).
MLCrosstalk is an advanced statistical model based on Latent Dirichlet Allocation (LDA) that links multiple data types to build the entire interactome for SARS-CoV-2. MLCrosstalk can integrate samples with multiple information layers, ensure a consistent topic distribution on all types of data, and deduce individual-level linkages that can differ between patients.
The researchers also implemented a secondary refinement with network propagation to enable the microbe-gene linkages to focus on larger network structures. They first evaluated the trained model by analyzing the clustering of sample topic distributions. The model groups individuals with the coronavirus disease 2019 (COVID-19), healthy individuals, and those with community-acquired pneumonia (CAP) into distinct clusters.
The authors used multiple known gene sets such as the Kyoto Encyclopedia of Genes and Genomes (KEGG), virus-host protein-protein interaction (PPI), WikiPathways, and COVID-19-related gene sets to annotate the functions of top-weighted genes.
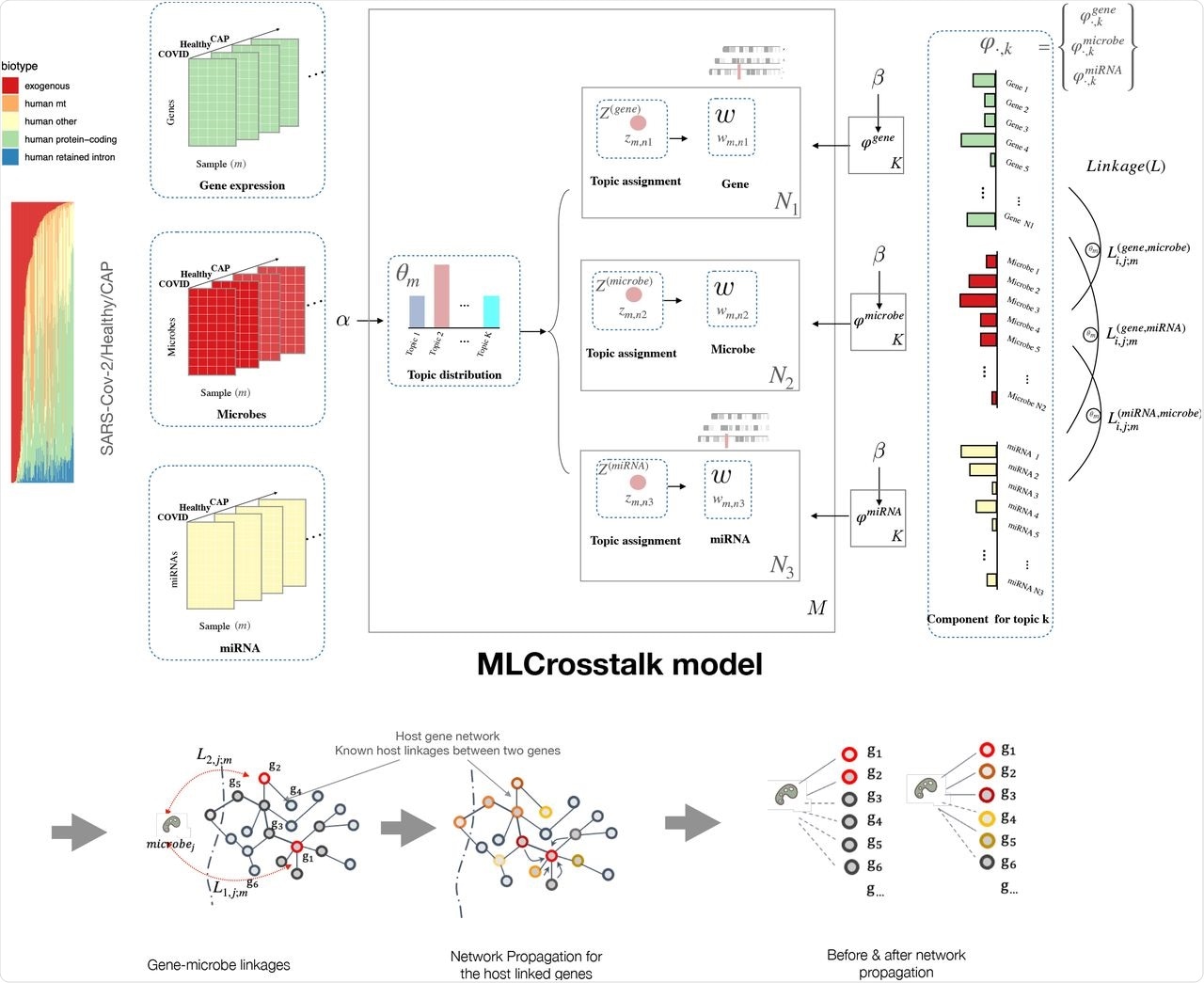 MLCrosstalk workflow. We transform gene expression, microbe abundance, and (pre)miRNA expression data, which are then input into the MLCrosstalk model. After training, we apply network propagation to refine the linkages. Multiple layer comparison and network tracing can identify shared and specific pathways and connections.
MLCrosstalk workflow. We transform gene expression, microbe abundance, and (pre)miRNA expression data, which are then input into the MLCrosstalk model. After training, we apply network propagation to refine the linkages. Multiple layer comparison and network tracing can identify shared and specific pathways and connections.
Study findings
The MLCrosstalk model had three major advantages in the integration analysis of multiple data types. To this end, this modeling approach handled sparse and noisy data using the Dirichlet distribution of hyperparameter, unifies topic distribution for all patient/samples, thus facilitating linkage identification across various types of data, and can be easily extended to many data types with missing samples.
Upon analysis of COVID-19 datasets, MLCrosstalk extracted dimensionally reduced patterns to show a detailed link between host protein-coding genes, non-coding genes, and microbes. MLCrosstalk constructed a comprehensive interactome for the gene-microbe-miRNA network, which was further refined through network propagation to integrate pathway data and link host-pathogen interactions with biological relevance.
The researchers used the Kullback-Leibler divergence between topic distributions and compared it to a random background to find that Topic 9 is the most interesting topic and differed this from the background distribution. Using multiple known gene sets, the authors annotated the functions of the top-weighted genes part of Topic 9 to discover that these genes are highly enriched in heat-shock response proteins and immune-related pathways.
The researchers analyzed and compared microbes with possible associations with SARS-CoV-2 and found that Rothia mucilaginosa, Prevotella melaninogenica, and Haemophilus parainfluenzae showed reduced relative abundance in patients with COVID-19. The results also showed that genes involved in the Notch signaling pathway like NOTCH4, HDAC2, PSEN1 are upregulated significantly in the lungs during COVID-19. Other bacteria like Escherichia coli, Staphylococcus aureus, and Klebsiella pneumoniae were also found to be highly associated with COVID-19, although many of these, especially gram-negative bacteria, could be nosocomial in some contexts.
The researchers inferred COVID-19-specific linked genes by comparing their occurrences in healthy individuals and those with COVID-19. They found that VEGFA-VEGFR2 and cytoplasmic ribosomal protein was associated with COVID-19.
Top-ranked pathways were identified using a random walk with restart (RWR) approach, whierein the findings identified the VEGFA-VEGFR2 as well as the immune pathway. MLCrosstalk identified genes like IFNAR1, IFNAR2, and STAT associated with the viral entry.
Conclusions
The MLCrosstalk statistical model developed by the researchers overcame three challenges including heterogeneity and noise in data, integration of multiple data types, and personalized linkage identification. Using MLCrosstalk, a list of genes and microbes associated with SARS-CoV-2 were identified, latent patterns of multiple data sets were retrieved, and sample-specific linkages showing biological evidence were identified.
The team identified the microbial co-infections associated with COVID-19, with some microbes showing synergistic and antagonistic effects with COVID-19. The linked genes of R. mucilaginosa had a high representation in COVID-19 patients, while those of P. melaninogenica had a lower representation in the aforementioned pathway creating an opposite pattern. Such distinct patterns were also seen between these two microbe groups for other pathways like immune-response, type II interferon signaling, and Notch signaling pathways.
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Lou, S., Li, T., & Gerstein, M. (2021). Constructing a multiple-layer interactome for SARS-CoV-2 in the context of lung disease: Linking the virus with human genes and co-infecting microbes. bioRxiv. doi:10.1101/2021.12.05.471290. https://www.biorxiv.org/content/10.1101/2021.12.05.471290v1.
Posted in: Genomics | Medical Science News | Medical Research News | Disease/Infection News
Tags: Bacteria, Coronavirus, Coronavirus Disease COVID-19, Gene, Gene Expression, Genes, Healthcare, heat, Interactome, Interferon, Lung Disease, Lungs, micro, Notch Signaling, Pathogen, Pneumonia, Propagation, Protein, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Signaling Pathway, Staphylococcus aureus, Syndrome, Therapeutics, Virus

Written by
Susha Cheriyedath
Susha has a Bachelor of Science (B.Sc.) degree in Chemistry and Master of Science (M.Sc) degree in Biochemistry from the University of Calicut, India. She always had a keen interest in medical and health science. As part of her masters degree, she specialized in Biochemistry, with an emphasis on Microbiology, Physiology, Biotechnology, and Nutrition. In her spare time, she loves to cook up a storm in the kitchen with her super-messy baking experiments.
Source: Read Full Article