
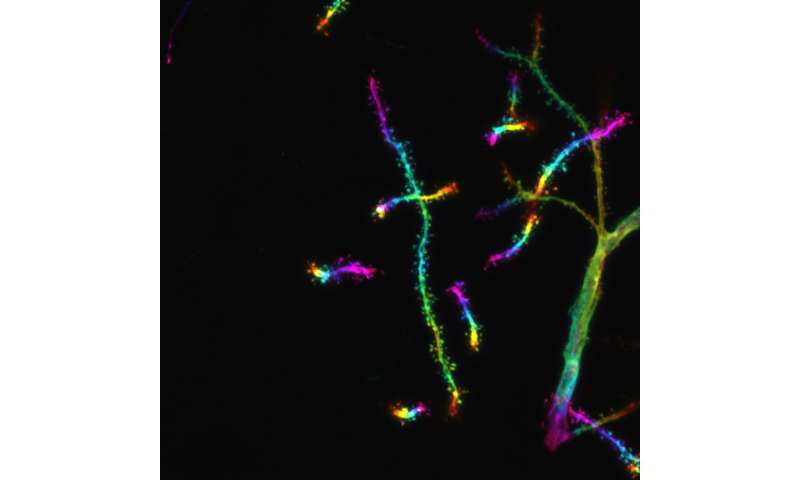
, an indicator of silenced heterochromatin. Credit: ©Hagelkruys/IMBA")
The gene-silencing complex HUSH might be involved in complex disorders affecting the brain and neurons. However, its mechanism of action remains unclear. Researchers from the Institute of Molecular Biotechnology of the Austrian Academy of Sciences (IMBA) now uncover the in vivo targets and physiological functions of a component of the HUSH gene-silencing complex and one of its associated proteins.
The work, conducted in laboratory mouse models and human brain organoids, links the HUSH complex to normal brain development, neuronal individuality and connectivity, as well as mouse behavior. The findings are published in Science Advances.
The human silencing hub (HUSH) complex was recently identified to be of key importance for silencing repetitive genetic elements including transposons in mammals. The HUSH complex contains MPP8, a protein that binds the histone modification mark H3K9me3. Additionally, HUSH is known to recruit other proteins including the zinc finger protein MORC2.
In humans, mutations affecting MORC2 are associated with axonal neuropathy, a type of nerve damage, and neurodevelopmental disorders. However, little is known about the physiological functions of MPP8 and MORC2 or how they might affect brain health. Researchers led by Astrid Hagelkruys, Senior Research Associate in the Penninger group at IMBA, therefore set out to investigate the targets and functions of these two proteins in vivo in laboratory mouse models and in human brain organoids.
Altered brain development and behavior
The team used a comprehensive in vivo approach including behavioral, motor, developmental, genetic, and transcriptomic experiments. They found that MPP8 and MORC2A (the mouse ortholog of human MORC2) were highly expressed in the brain, where they are exclusively found in neurons. “We demonstrated that MPP8 and MORC2A play a role in normal brain development, the specification of neuronal identity and connectivity of neurons, as well as mouse behavior,” says Astrid Hagelkruys, the project lead and co-corresponding author of the study.
Furthermore, deleting MPP8 or MORC2A in the nervous system of the mouse models increased brain size and altered brain architecture without major changes in transposable element expression. These deletions affected the mice’s motoric functions and behavior. “Hence, surprisingly in a living animal, we showed that MPP8 and MORC2A act beyond transposable element regulation,” says Hagelkruys.
Molecular mechanism reminiscent of gene silencing
So far, the HUSH complex has been involved in transposon regulation. “We showed that MPP8 and MORC2A suppressed the protocadherin gene clusters in an H3K9me3-dependent manner. At the protein level, these protocadherin gene clusters form neuronal surface proteins that mediate contact with other neurons. Although protocadherins are not transposable elements, some are expressed in the central nervous system as ‘repetitive-like’ gene clusters,” explains Hagelkruys.
In the mouse models, MPP8 and MORC2A specifically silenced the protocadherin cluster on mouse chromosome 18. Deleting MPP8 and MORC2A led to more synapses forming in the neurons, which may coincide with an impairment of neuronal individuality. In other words, the ability of neurons to distinguish “self” from “non-self.” By expressing different combinations of clustered protocadherins, neurons acquire a form of “barcode” that allows them to control the formation of synaptic connections with other neurons. Hence, by targeting clustered protocadherins, MPP8 and MORC2A may ensure that neurons acquire the right “barcode” and form synapses only with the correct counterparts.
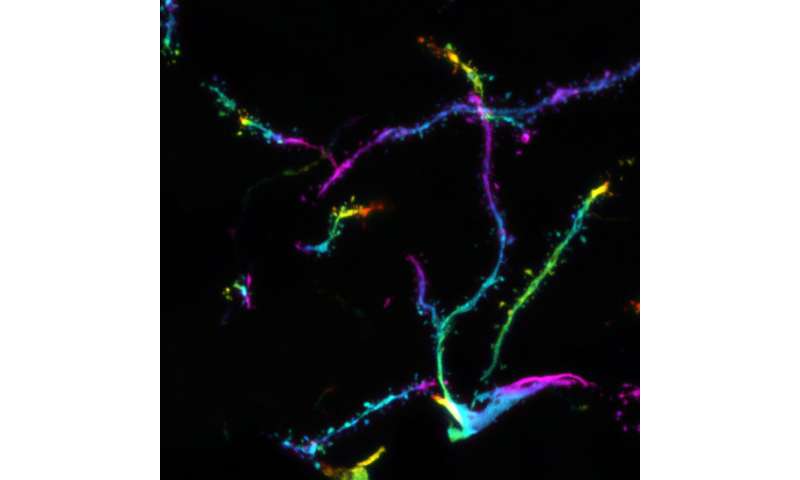
In addition, the team examined the effects of MPP8 and MORC2 deficiency in human brain organoids. Using this stem cell-derived model of the human brain, the scientists observed concordant results: the absence of MPP8 or MORC2 led to increased numbers of clustered protocadherins expressed in organoid neurons at the single-cell level. This indicates that the absence of the two proteins disrupted neuronal identity also in the human brain organoids.
The epigenetics of brain and nervous system diseases
With the current work, the researchers uncovered a pivotal role of the HUSH complex in the epigenetic regulation of protocadherin expression in the nervous system. These findings link the mechanistic effect of suppressing repetitive-like genetic elements with mouse brain physiology and behavior. The team’s brain organoid results also show that similar effects may be found in humans.
“The interest of these findings on the essential function of the HUSH complex in the brain lies in the implication of protocadherins in neuronal fidelity and brain evolution. However, how this is regulated remained largely unknown. The dysregulation of clustered protocadherins has been associated with various neurological and neurodevelopmental diseases, but also multiple mental disorders in humans. Hence, our findings might help us better understand the epigenetic regulation mechanisms governing these diseases and provide a new way to study brain evolution,” concludes Josef Penninger, group leader at IMBA.
More information:
Astrid Hagelkruys et al, The HUSH complex controls brain architecture and protocadherin fidelity, Science Advances (2022). DOI: 10.1126/sciadv.abo7247. www.science.org/doi/10.1126/sciadv.abo7247
Journal information:
Science Advances
Source: Read Full Article